
Keď sa hovorí o tom, čo dieťa zdedilo po jednotlivých členoch rodiny, najmä teda po rodičoch či starých rodičoch, najčastejšie sa spomínajú dobré a zlé vlastnosti či rozličné črty tváre a proporcie tela, napríklad veľký nos, plné pery, zelené oči, ale aj farba či hustota vlasov a podobne. V našich génoch je zapísané všeličo a, žiaľ, genetika môže mať vplyv aj na rozvoj rozličných ochorení.
Menovať by sme mohli napríklad celý zoznam dedičných chorôb, ktorých vznik je podmienený nejakou mutáciou, ktorá býva na ďalšiu generáciu spravidla prenesená rodičovskou zárodočnou bunkou, čiže spermiou alebo vajíčkom. Medzi takéto ochorenia radíme i takzvanú fenylketonúriu. Čo je fenylketonúria, kedy ju dieťa môže zdediť aj aké sú možnosti jej liečby, to vám prezradíme v nasledujúcom článku.
Čo je fenylketonúria?
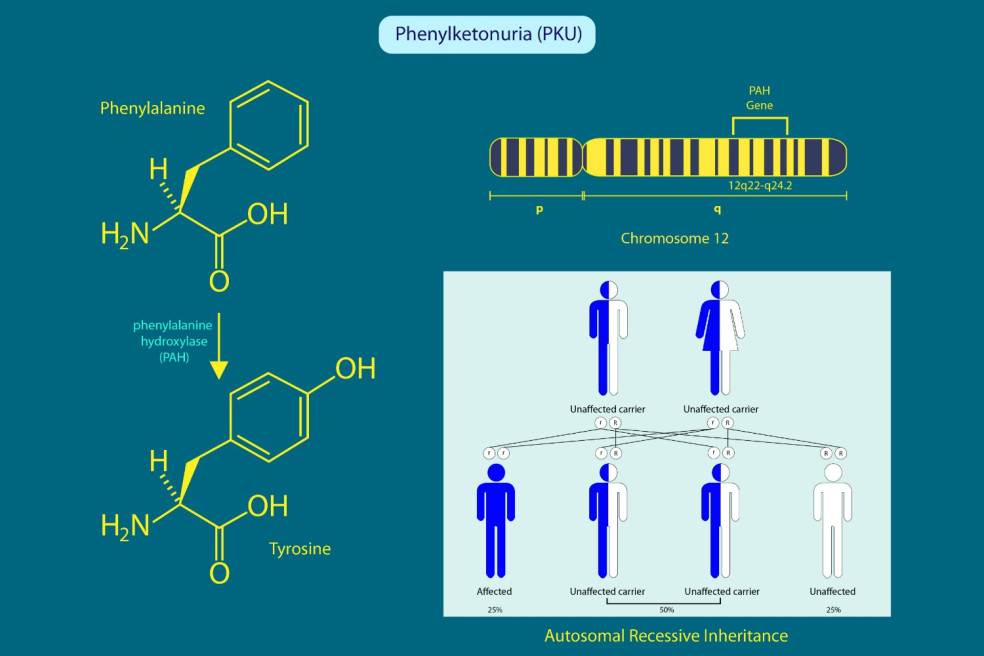
Pod pojmom fenylketonúria rozumieme metabolickú chorobu, ktorá je dedičná. Toto ochorenie je tiež známe pod skratkou PKU. Charakteristické je nedostatočnou činnosťou enzýmu zvaného fenylalanínhydroxyláza, ktorý je nevyhnutný pre rozklad aminokyseliny fenylalanín a jej premeny na aminokyselinu tyrozín. Hromadenie fenylalanínu v tkanivách a krvi následne zapríčiňuje rozvoj rozličných zdravotných ťažkostí.
Kedže je fenylketonúria dedičným ochorením, jej príčinu vzniku môžeme hľadať v mutácii génu pre spomínaný enzým, ktorý v zdravom organizme premieňa fenylalanín na tyrozín. Ten je umiestnený na 12. chromozóme. U postihnutých jedincov je tohto enzýmu nedostatok alebo úplne chýba.
Fenylketonúria je autozomálne recesívnym ochorením. To znamená, že dedičnosť nie je viazaná na pohlavie a ochorenie sa prejaví v prípade, že jedinec zdedí poškodený gén od oboch rodičov. Ak sú obaja rodičia nositeľmi génu, existuje 25 % riziko, že ich dieťa bude trpieť fenylketonúriou a zároveň 25 % pravdepodobnosť, že dieťa bude úplne zdravé. Vo zvyšných 50-tich percentách býva dieťa iba prenášačom génu.
V takomto prípade potomok získava od jedného rodiča zdravý gén, ktorý dokáže prekryť chybný gén od druhého rodiča a ochorenie sa tak neprejaví. Fenylketonúria sa vyskytuje v priemere u jedného zo 6000 až 10 000 novorodencov, záleží od konkrétnej krajiny či oblasti.
Odporúčame: Nadmerný smäd – nedostatok tekutín alebo príznak ochorenia? Pozor, môže to byť vážne!
Čo môže spôsobiť nadbytok fenylalanínu v krvi?
Ako sme spomenuli už vyššie, u jedincov s fenylketonúriou je hladina fenylalanínu zvýšená. Aminokyselina sa ukladá v krvi a tkanivách, a to v dôsledku narušenia jej metabolizmu, pri ktorom by s pomocou fenylalanínhydroxylázy dochádzalo k jej premene na aminokyselinu tyrozín. Tyrozín je nevyhnutný pre tvorbu ďalších látok, akými sú napríklad dopamín, adrenalín aj noradrenalín. Kvôli genetickej poruche si ho telo nevie správne produkovať.
Následkom týchto chybných procesov, respektíve nesprávnej funkcie metabolizmu sa u jedinca rozvíjajú viaceré, spravidla veľmi vážne zdravotné komplikácie. To nastáva v prípade, ak ochorenie nie je diagnostikované a nezačne sa s jeho liečbou. Fenylalanín nahromadený v tkanivách poškodzuje nervové vlákna a mozog a má negatívny vplyv na vývoj centrálnej nervovej sústavy.
Neliečená fenylketonúria spôsobuje mentálnu retardáciu aj oneskorený vývoj reči. Časté sú i poruchy správania a zníženie inteligenčných funkcií. Ďalej sa v dôsledku ochorenia vyskytujú i kožné problémy, stuhnutosť svalstva, spomalený rast a nižší vzrast, zmenšenie mozgovej časti hlavy aj slabá pigmentácia pokožky či vlasov.
Moč postihnutého zapácha, pretože sa ním sčasti vylučuje neodbúraný fenylalanín. Ochorenie sa u novorodenca rozvíja po tom, ako začína prijímať mlieko, pričom už po pár mesiacoch sa začína fenylalanín hromadiť a prichádza k výskytu prvých ťažkostí. Už medzi 6. a 12. mesiacom veku jedinca dochádza k výraznému prejavovaniu ochorenia pacienta.
Je možné vyliečenie?
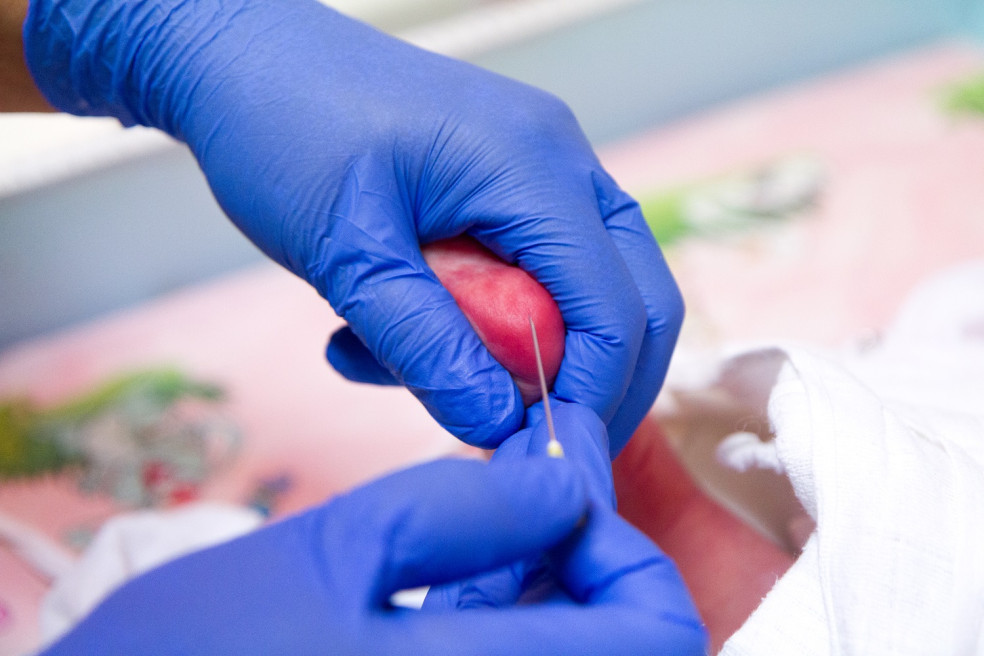
Pre diagnostiku fenylketonúrie, ale aj ďalších ochorení, sa v súčasnosti u nás vykonáva celoplošný novorodenecký skríning, počas ktorého sa testuje krv odobraná z päty dieťaťa. Pri zistení poruchy enzýmu fenylalanínhydroxyláza sa pre potvrdenie diagnózy pristupuje ešte k ďalším genetickým vyšetreniam. Po potvrdení ochorenia je nevyhnutné okamžite začať s liečbou, aby sa tak zabránilo poškodeniu centrálnej nervovej sústavy.
Fenylketonúria, žiaľ, sprevádza jedinca počas celého života. Zdedený chybný gén pre spomínaný enzým totiž nie je možné v súčasnosti nejako napraviť. Liečba spočíva primárne v úprave jedálnička, pričom je nevyhnutná strava s obmedzeným množstvom aminokyseliny fenylalanín. Pri skorom nasadení celoživotnej diéty sa dieťa vyvíja normálne. Konkrétny stravovací plán určuje lekár na základe individuálnych požiadaviek pacienta, pričom sa zohľadňujú mnohé parametre.
Väčšie obmedzenia majú deti či tehotné ženy s týmto ochorením, naopak v dospelosti je za bežných okolností diéta voľnejšia. Pacienti sa musia vyhýbať jedlám s vysokým obsahom fenylalanínu, akými sú napríklad vajcia, mäso, strukoviny, čokoláda či syry. O niečo nižší podiel spomínanej aminokyseliny sa nachádza v kukurici, zemiakoch, špenáte či kapuste.
Keďže je prísun bielkovín, respektíve aminokyselín pre zdravie človeka zásadný, jedincom s týmto ochorením sú podávané špeciálne zmesi aminokyselín s minerálmi a vitamínmi, ktoré fenylalanín neobsahujú. Telu je tiež dôležité dodávať dostatočné dávky tyrozínu, ktorého produkcia je v tele v dôsledku ochorenia narušená alebo úplne zastavená. U pacientov by rovnako mala byť pravidelne kontrolovaná hladina fenylalanínu v krvi.
Pozri aj: Choroby prenosné zo zvieraťa na človeka: Pri tejto si dávaj obzvlášť pozor!











