
Už ste u nás mali možnosť dozvedieť sa množstvo zaujímavých a dôležitých informácií o rozličných dedičných ochoreniach a rozšíriť si tak znalosti i v tejto oblasti. Venovali sme tomu, ako sa prejavuje Kartagenerov syndróm, čo je Huntingtonova choroba, čo spôsobuje Downov syndróm a aj tomu, kedy sa narodí človek s albinizmom.
Dnes sa pozrieme na to, ako vzniká závažná a dedičná cystická fibróza, ktorá výrazne poškodzuje najmä orgány dýchacej sústavy, no ktorá môže postihnúť i tráviaci trakt človeka. Zistite, ako sa toto ochorenie prejavuje a aké sú v súčasnosti možnosti liečby.

Čo je cystická fibróza?
Pod pojmom cystická fibróza rozumieme závažné genetické multisystémové ochorenie, ktoré najčastejšie postihuje dýchací a tráviaci systém človeka, niekedy však aj iné orgány. Keďže ide o dedičné ochorenie, vzniká na základe mutácie génu, a to v tomto prípade konkrétne génu pre CFTR, ktorý sa nachádza na dlhom ramienku 7. chromozómu. Ten má význam pri udržiavaní vnútorného prostredia bunky.
Reč je o bielkovinovej štruktúre, ktorá funguje ako chloridový kanál s regulačnou funkciou. Pri mutácii tohto génu dochádza k narušeniu prenosu iontov a solí cez bunkovú membránu, čo spôsobuje zdravotné ťažkosti, respektíve sprievodné symptómy ochorenia.

Dedičnosť pri cystickej fibróze
Cystická fibróza je autozomálne recesívne dedičné ochorenie. To znamená, že na jeho rozvoj u jedinca je nevyhnutné, aby zdedil poškodený gén od oboch rodičov. V prípade, že človek zdedí jeden zdravý gén, choroba sa u neho nerozvinie, no stáva sa z neho nositeľ defektného génu, ktorý môže opäť preniesť na svoje potomstvo.
Aj keď sú obaja rodičia prenášači, je tu taktiež šanca, že sa im narodení zdravé dieťa, ktoré poškodený gén nezdedilo ani po jednom z nich, čiže ho nemôže ani prenášať ďalej. Cystická fibróza je najmä v Európe pomerne rozšíreným genetickým ochorením. V priemere sa objavuje jeden prípad na 3000 novorodencov.
Ak sú obaja rodičia prenášačmi génu pre cystickú fibrózu:
25 % šanca, že dieťa zdedí dva poškodené gény → rozvinie sa u neho cystická fibróza.
50 % šanca, že dieťa zdedí jeden zdravý a jeden poškodený gén → bude zdravým prenášačom, ochorenie sa u neho neprejaví, no môže gén ďalej preniesť.
25 % šanca, že dieťa zdedí dva zdravé gény → bude úplne zdravé a nebude ani prenášačom.
Ak je prenášač len jeden rodič, riziko, že dieťa ochorie, je nulové. Dieťa môže byť iba prenášačom (50 % šanca), alebo úplne zdravé (50 % šanca).
Shutterstock®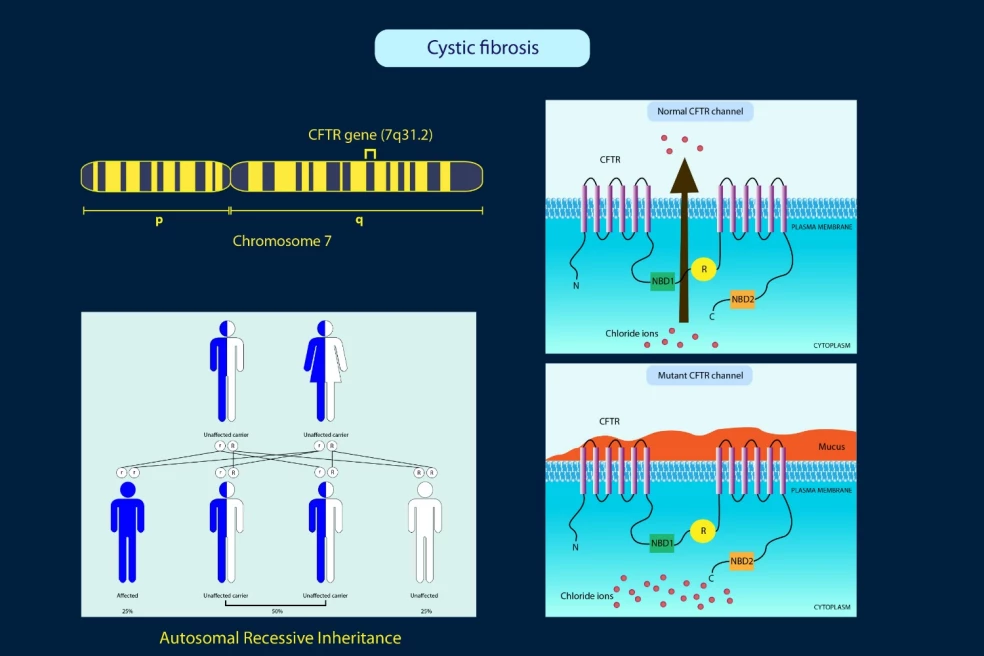
Ako sa cystická fibróza prejavuje?
V dôsledku mutácie spomínaného génu a narušenia jeho funkcie dochádza v tele postihnutého okrem iného najmä k výraznému zahusteniu hlienu a ďalších tekutín, ktoré sú tvorené v mnohých častiach tela. Práve toto zahustenie hlienu, ktorý v takomto prípade predstavuje dokonalé miesto pre usídlenie baktérií a nečistôt, je zodpovedné za väčšinu klinických prejavov cystickej fibrózy.
Príliš hustý sekrét prispieva k upchávaniu dýchacích ciest a množeniu baktérií. Taktiež bývajú postihnuté orgány tráviacej sústavy, a to najmä pankreas či žlčník, čo spôsobuje nedostatočné vylučovanie tráviacich enzýmov a žlče. U mužov môžu byť zasiahnuté napríklad i semenovody.
Nástup príznakov cystickej fibrózy v detstve
Prejavy cystickej fibrózy postupne nastupujú už od narodenia jedinca, a to neraz v priebehu niekoľkých mesiacov, prípadne počas prvých rokov. U detí je charakteristické v prvom rade celkové neprospievanie, spomalený rast, nepriberanie na váhe až chudnutie, a to i napriek dostatočnej výžive. Typickým prejavom je aj veľmi slaný pot.
Respiračné a tráviace problémy pri cystickej fibróze
Okrem toho sa u jedincov s cystickou fibrózou objavujú najmä respiračné a tráviace ťažkosti. Často jedinca trápi dychová nedostatočnosť a vlhký neutíchajúci kašeľ, pričom postihnutý vykašliava hustý hlien. Bežné sú aj infekcie dýchacích ciest. U pacienta sa opakovane môžu vyskytovať zápaly prínosných dutín, priedušiek aj pľúc s bolesťou v oblasti hrudnej kosti, vysokými teplotami a dráždivým kašľom.
Pri postihnutí tráviacej sústavy často dochádza najmä k hromadeniu hlienov v pankrease, respektíve jeho vývodoch. Následne tak býva u jedincov narušená funkcia trávenia, čím dochádza i k obmedzenému vstrebávaniu zložiek potravy. Pacient tak môže trpieť na nedostatok mnohých dôležitých látok. Okrem toho sa pridružujú i hnačky, bolesti a kŕče v bruchu. Prejavom cystickej fibrózy u mužov býva i neplodnosť.
Možné komplikácie cystickej fibrózy
chronické infekcie pľúc a postupné zhoršovanie pľúcnych funkcií,
bronchiektázie (trvalé rozšírenie priedušiek),
vznik diabetes mellitus spojeného s cystickou fibrózou,
cirhóza pečene alebo iné poruchy pečeňových funkcií,
neplodnosť u mužov, problémy s plodnosťou aj u žien,
osteoporóza a časté zlomeniny pre zníženú hustotu kostí.

Diagnostika cystickej fibrózy
Cystická fibróza sa dá diagnostikovať viacerými spôsobmi. Najčastejšie sa u detí využíva novorodenecký skríning, ktorý je na Slovensku súčasťou štandardnej starostlivosti. Ak test odhalí možné riziko, nasleduje tzv. potný test, pri ktorom sa meria koncentrácia chloridov v pote – ich nadmerná hodnota je typickým znakom ochorenia. V prípade nejasností sa pristupuje k genetickým testom, ktoré dokážu potvrdiť mutáciu v CFTR géne.
Diagnostika sa môže doplniť aj o funkčné vyšetrenia pľúc, tráviaceho systému alebo pečene, aby sa zistilo, do akej miery je organizmus ochorením zasiahnutý. Včasné odhalenie je mimoriadne dôležité – umožňuje totiž začať s liečbou a rehabilitáciou už od prvých mesiacov života dieťaťa, čím sa výrazne zlepšujú prognózy a kvalita života.
Shutterstock®
Je možné CF odhaliť už počas tehotenstva?
Pri niektorých prípadoch je možné cystickú fibrózu diagnostikovať ešte pred narodením dieťaťa. Ak je známe, že rodičia sú nositeľmi mutácie CFTR génu, môže sa vykonať prenatálne genetické testovanie. To zahŕňa vyšetrenia ako amniocentéza (odber plodovej vody) alebo choriová biopsia (odber vzorky placenty), ktoré umožňujú analyzovať DNA plodu.
V posledných rokoch sa využívajú aj neinvazívne prenatálne testy (NIPT), ktoré sa robia z krvi matky. Ide o platené vyšetrenia, ktoré dokážu s vysokou presnosťou zachytiť niektoré chromozómové abnormality a pri rozšírených verziách aj vybrané genetické ochorenia vrátane cystickej fibrózy. Výhodou je, že sú bezpečné pre matku aj dieťa, keďže nevyžadujú zásah do tela.
Okrem toho existujú aj metódy tzv. preimplantačnej genetickej diagnostiky, ktoré sa využívajú pri umelom oplodnení. Embryá sa v laboratóriu vyšetrujú ešte pred vložením do maternice a vyberajú sa tie, ktoré nemajú poškodený gén. Prenatálna diagnostika však prináša aj etické otázky a rozhodnutie vždy zostáva na rodičoch po konzultácii s genetikom.

Je cystická fibróza liečiteľná?
Cystická fibróza je, podobne ako iné genetické ochorenia, nevyliečiteľná. Ani moderná medicína nedokáže nejakým spôsobom opraviť poškodený gén, ktorý jedinec zdedil. Liečba je však nevyhnutná, keďže pri tomto ochorení sa môžu rozvinúť mnohé komplikácie, ktoré vedú i k smrti pacienta. Najmä opakované infekcie respiračného systému môžu prispieť k zlyhaniu pľúc.
Podporná liečba cystickej fibrózy
Liečba cystickej fibrózy spravidla prebieha v špecializovaných centrách. Niekoľko zariadení toho druhu máme i na Slovensku. Liečba spočíva v udržiavaní čo najlepšieho zdravotného stavu pacienta a prevencii vážnych infekcií a iných komplikácií. Zahŕňa užívanie antibiotík aj rôzne dychové rehabilitácie a využívanie špeciálnych techník na vykašliavanie hustého hlienu.
Pacientom sú často podávané formou tabletiek aj chýbajúce tráviace enzýmy, pričom zvyčajne bývajú na vysokokalorickej strave s vysokým podielom tukov. Okrem toho je nevyhnutná liečba komplikácií, ktoré sa u konkrétneho pacienta vyskytnú, čiže hlavne rozličných tráviacich a respiračných ťažkostí. V pokročilých štádiách ochorenia, ak je to potrebné a hlavne možné, sa pristupuje aj k transplantácii pľúc.
Prognózy pri cystickej fibróze
Dôsledná liečba cystickej fibrózy môže skvalitniť a predĺžiť život pacienta. Priemerná dĺžka pacientov s touto chorobu stále rastie. Hoci sa však jedinci dožívajú v priemere 35 až 40 rokov a v niektorých krajinách dokonca i 50, v mnohých krajinách nemalý počet chorých podľahne ochoreniu už v detskom veku, a to často bez diagnostikovania choroby.
Život s cystickou fibrózou
Život pacienta s cystickou fibrózou je náročný a vyžaduje si každodennú starostlivosť, no vďaka modernej medicíne dnes ľudia s týmto ochorením žijú dlhšie a kvalitnejšie než v minulosti. Pacienti musia pravidelne absolvovať inhalácie a dychové cvičenia na uvoľnenie hlienu, užívať antibiotiká pri infekciách či dopĺňať tráviace enzýmy v podobe tabletiek. Dôležitá je aj výživa – odporúča sa vysokokalorická strava s vyšším obsahom tukov a vitamínov, aby sa predišlo podvýžive.
Veľkú úlohu zohráva aj psychická podpora a zapojenie rodiny. Deti i dospelí s CF sa často stretávajú s obmedzeniami, no napriek tomu môžu viesť plnohodnotný život – chodiť do školy, športovať a venovať sa svojim záľubám. Moderné lieky, najmä tzv. CFTR modulátory, dnes zásadne menia prognózy pacientov a dávajú nádej na dlhší a aktívnejší život.
Nádej v medicínskom pokroku
Cystická fibróza je síce nevyliečiteľné genetické ochorenie, no včasná diagnostika, podporná liečba a nové terapie dokážu výrazne predĺžiť život pacientov a zlepšiť jeho kvalitu. Hoci ochorenie prináša množstvo výziev, starostlivosť lekárov, rodiny a moderné lieky otvárajú pacientom cestu k plnohodnotnému detstvu aj dospelosti. Nádej spočíva v tom, že medicína napreduje a s ňou aj šance na lepší život ľudí s cystickou fibrózou.
Aktualizovaný článok: 22.09.2025









0 komentárov